Stereo-seq Input Tutorial
Required files
Filename / pattern |
Required |
Format |
Description |
|---|---|---|---|
|
At least one |
gef |
Cell-by-gene expression matrix across different bin sizes |
|
Required |
TIFF/OME.TIFF/OME-XML |
Contains TIFF image information |
|
No |
h5ad |
Pipeline analysis results |
For Stereo-seq, we require that the file layout follows the output directory structure produced by SAW V8, the analysis software developed by BGI (Stereo-seq platform).
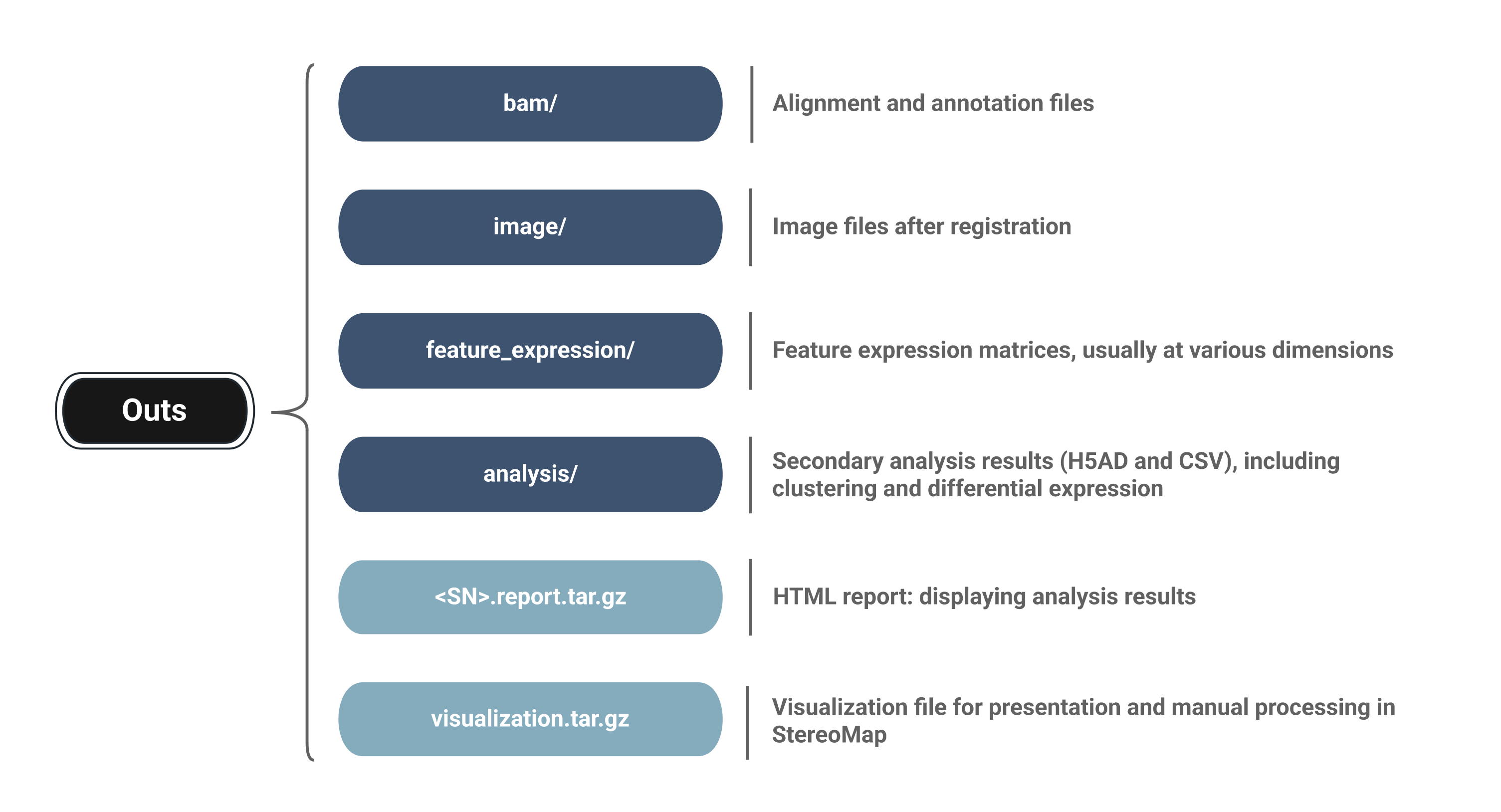
Through open community contributions to the SpatialData project, we have optimized the Stereo-seq reader to support SAW V8 format, and, based on contributions from @brainfo, added support for selecting cellbin and adjusted.cellbin data. The reader logic has been further refined for integration with Spatialsnake. We thank @brainfo for the contributed solution: spatialdata-io/stereoseq.py
The required directory layout is shown below:
project_root/
├── data/ (stores your raw data)
│ └── {sample_id}/
├── sample.txt (key sample description file)
└── results/ (stores analysis outputs)
data/
└── {sample_id}/
├── feature_expression *required at least
│ ├── {sample_id}.tissue.gef
│ ├── {sample_id}.cellbin.gef
│ ├── {sample_id}.adjusted.cellbin.gef
└── image/ *required at least
│ └── {sample_id}_HE_regist.tif
└── analysis/ *optional when load_analysis=False
├── {sample_id}.bin20_1.0.h5ad
└── {sample_id}.bin50_1.0.h5ad
Where these files come from
Official download: Public demonstration datasets provided on the BGI STOmics resource portal
Experimental output: Sample data obtained from the BGI STOmics Stereo-seq sequencing platform
Public dataset download: If you wish to use public datasets, please organize the required files according to the directory hierarchy described above for each sample you intend to analyze
Demo Walkthrough
run_type: stereo_seq. In this tutorial, we use a public Mouse Brain Demo Data and organize the downloaded files into the directory structure expected by Spatialsnake.
One convenient public source for the required processed files is:
STOmics Stereo-seq Demo Dataset
To keep the demo concise, we download only the required feature-expression files and the corresponding TIFF image file.
Example setup:
mkdir -p project_root/data/Mouse_Brain
cd project_root/data/Mouse_Brain
mkdir -p feature_expression image
cd feature_expression
curl -C - -O https://demo.stomicsdb.tech/C04042E2_Mouse_Whole_Brain_Stereo-seq_FF_V1.3_ssDNA/outs/C04042E2.adjusted.cellbin.gef
curl -C - -O https://demo.stomicsdb.tech/C04042E2_Mouse_Whole_Brain_Stereo-seq_FF_V1.3_ssDNA/outs/C04042E2.cellbin.gef
curl -C - -O https://demo.stomicsdb.tech/C04042E2_Mouse_Whole_Brain_Stereo-seq_FF_V1.3_ssDNA/outs/C04042E2.tissue.gef
cd ../image
curl -C - -O https://demo.stomicsdb.tech/C04042E2_Mouse_Whole_Brain_Stereo-seq_FF_V1.3_ssDNA/outs/C04042E2_ssDNA_regist.tif
cd ../
After download, the sample directory should match the layout shown below.
project_root/
├── data/ (stores your raw data)
│ └── Mouse_Brain/
├── sample.txt (key sample description file)
└── results/ (stores analysis outputs)
data/
└── Mouse_Brain/
├── feature_expression *required at least
│ ├── C04042E2.tissue.gef
│ ├── C04042E2.cellbin.gef
│ ├── C04042E2.adjusted.cellbin.gef
└── image/ *required at least
└── C04042E2_ssDNA_regist.tif
Input validation logic
In a standard spatial transcriptomics analysis pipeline, Spatialsnake ingests only one data format at a time: either a single bin size, or the SAW cell segmentation output (cellbin or adjusted.cellbin). This approach allows deeper exploration of the resolution level of interest while reducing memory usage. As with the bin selection strategy for Visium HD, we recommend choosing one bin size (e.g., 20 or 50) or one cell segmentation format (cellbin or adjusted.cellbin) and entering that value in sample.txt.
Multiple formats can also be specified; if needed, separate them with commas.
We demonstrate with the cellbin result. Copy the first example below and manually enter it into sample.txt.
single_analysis:
sample_id input_path bin_size
Mouse_Brain data/Mouse_Brain cellbin
Multiple bin sizes:
sample_id input_path bin_size
Mouse_Brain data/Mouse_Brain 20,50
We provide only the minimal command here. Please modify additional parameters as needed for your analysis.
spatialsnake single_analysis sample.txt stereo-seq --option=integrate
Output structure after ingestion
results/
├── Mouse_Brain/
└── integrate/
├── Mouse_Brain.zarr
├── total.png
├── total_umi_by_sample.png
├── total_genes_by_sample.png
├── genes_by_sample.png
└── scatter.png
Main output:
results/<sample>/integrate/<sample>.zarrAdditional output for comparison analysis:
results/merge_data/integrate/concatenated_sdata.zarrAdditional QC plots: the ingestion script writes five QC figures into the
integratedirectory. These files are generated during execution even though they are not explicitly listed in the Snakemakeoutputdeclaration.
You have now ingested your data into a zarr object. For the subsequent core analysis, please refer to Core Analysis Workflow. We recommend starting with the example dataset to gain hands-on experience with the basic core-analysis workflow. If you prefer to proceed directly with your own data, each step page begins with a concise summary of the essential parameters.
Simply follow the tutorial to update the sample name and platform-specific parameters, then continue with the next step: Preprocessing.
If you want to run multi-sample integration analysis, continue to Spatialsnake for multi-sample integration.