Reannotation (reannotation)
reannotation is the reannotation branch under annotation. It remaps celltype labels based on existing clusters or recluster labels.
Compared with manual, reannotation is designed for quickly revising existing labels and exporting the updated annotations in a standardized format, making it well suited to small naming adjustments after clustering has stabilized.
For the configuration reference, see annotation.yaml Reference.
Workflow overview
Read the downstream object listed in the second column of
sample.txt(.zarror.h5ad).Read the annotation mapping file and parse the relationship from
clusterorreclustertocelltype.Use
obs['recluster']if it exists; otherwise fall back toobs['clusters'].Write the mapped result to
obs['celltype']and assignUnknownto unmatched labels.Export the reannotated object together with
celltype_annotations.csvfor downstream analysis or delivery.
Note
We recommend completing clustering or reclustering and confirming that the cluster structure is stable before running reannotation. If the clustering itself is unreliable, reannotation will only amplify the existing bias.
Because this module repeats the step of integrating annotation information, we use the example data directly here. For other scenarios, adjust the required parameters as appropriate.
Prepare sample.txt and the mapping file annotation.txt
samples path_to_dir
Colon_Cancer_P2_008um results/Colon_Cancer_P2_008um/reclustering/Colon_Cancer_P2_008um.zarr
The current implementation skips the first line and reads the second line as the mapping definition. The second line is comma-separated, and the order corresponds to cluster or recluster IDs 0,1,2....
celltype
Tumor_I,Tumor_II,Tumor_III,Tumor_IV,Tumor_IV
In this example, the mapping is 0->Tumor_I, 1->Tumor_II, 2->Tumor_III, 3->Tumor_IV, and 4->Tumor_IV. If more IDs are present, continue listing them on the same line.
Run the command
spatialsnake single_analysis sample.txt visium_HD --option=annotation --anno_algorithm=reannotation --annotation-file=annotation.txt
or
spatialsnake single_analysis sample.txt visium --option=annotation --configfile annotation.yaml
Result file structure
This example shows single-sample visium reannotation. Before continuing to downstream analysis, first confirm that {sample}.zarr and celltype_annotations.csv have been generated.
results/
└── {sample}/
└── reannotation/
├── {sample}.zarr/ # for slide_seq, this becomes {sample}.h5ad
└── celltype_annotations.csv
The updated {sample}.zarr (or .h5ad) contains the revised obs['celltype'] field. celltype_annotations.csv is the standardized export table used for external review and data sharing.
Interpreting the results
In this example, the reclustered tumor population is reannotated into four distinct malignant subpopulations based on differences in their marker profiles.
The workflow also exports UMAP plots, spatial mapping figures, and cell-proportion plots after reannotation, allowing the revised labels to be evaluated from multiple perspectives.
A cell ID-to-celltype CSV file is also exported to support subsequent visualization and annotation transfer.
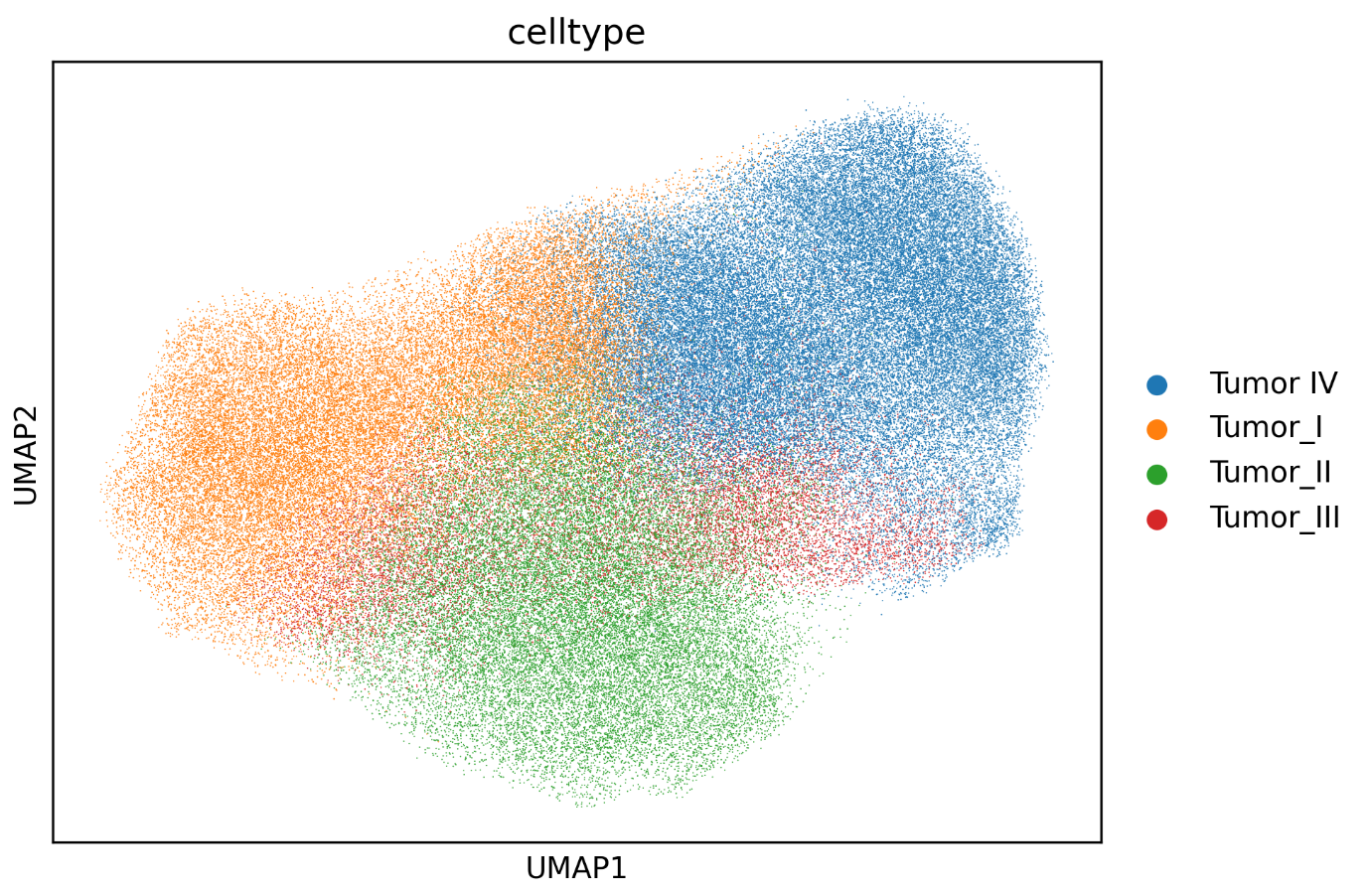
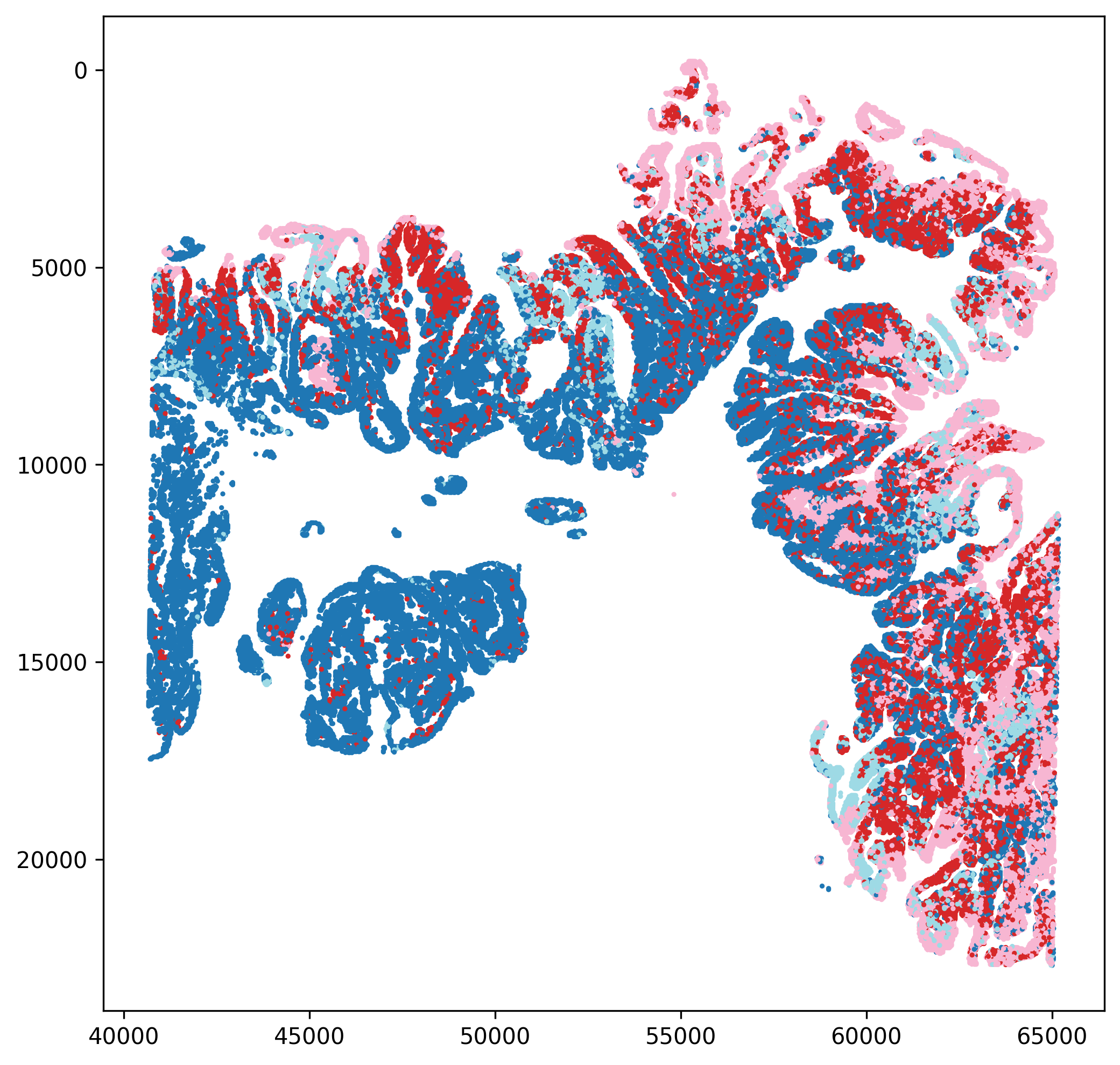
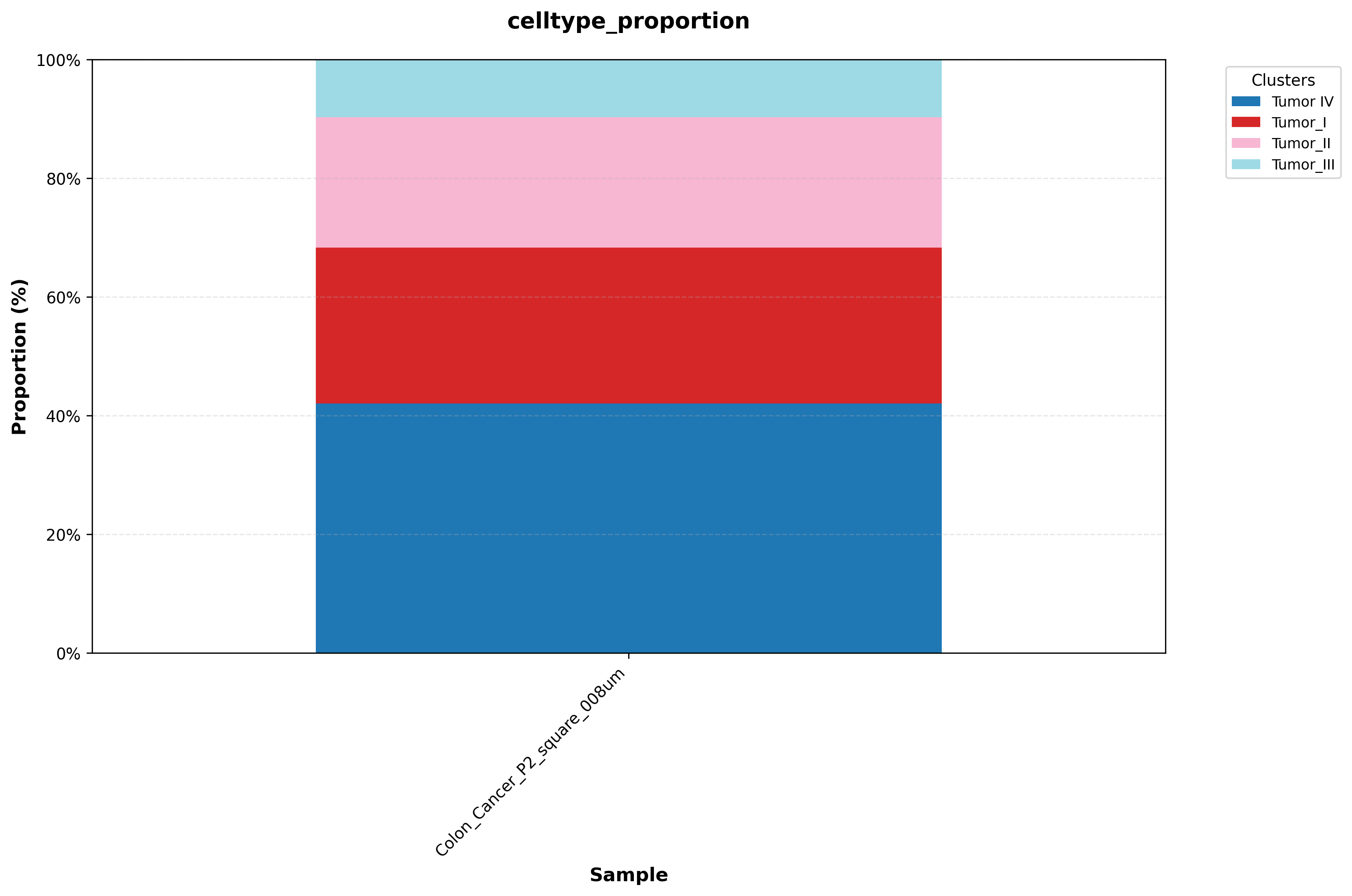
If you are building atlas-style annotations, you may need to annotate each subcluster in detail and then merge the results. Spatialsnake provides utility modules to make this process easier; see Utility Tools.
In the next step, we merge the annotated tumor subcluster labels back into the original larger dataset; see Merge Tool (merge).