Splitting Tool (splitting)
splitting is used to divide one spatial transcriptomics object into multiple smaller and easier-to-analyze subobjects.
Split by cell type or cluster, often for downstream subcluster analysis
Split by sample or experimental group, often for multi-sample organization before comparison
Split by ROI tables, often after lasso selection in Loupe or Xenium Explorer
Crop by image coordinates, often for focused local-region analysis
This page focuses on splitting zarr objects. For the configuration reference, see splitting.yaml Reference.
Typical use cases
You have completed
core_analysisand want to isolate one major cell class, such asTumor, for reclustering.You have an integrated multi-sample object and want to separate it by sample or group.
You selected ROIs in external software and want to import the CSV files for direct batch splitting.
You want to focus on a local tissue region, or one slide contains multiple samples that should be separated by coordinates.
Before you start
The input object should be a
.zarrpath, ideally fromintegrate,preprocess,clustering, orannotation.You know which field should be used for splitting, for example
celltype,clusters,sample, orgroup.The directory containing
sample.txtmatches your current working directory, or you use absolute paths in the command.
General command template
spatialsnake useful_tool --option=splitting <INPUT_ZARR_PATH> --split_by=<mode> --output_dir=results/useful_results
Scenario 1: split by cell type or cluster
Use this mode when you want to isolate one or more cell classes for further analysis, for example to refine the Tumor compartment in the example dataset.
spatialsnake useful_tool --option=splitting results/Colon_Cancer_P2_008um/annotation/Colon_Cancer_P2.zarr --split_by=celltype --barcodes=Tumor
You can also select multiple labels at once using commas, for example --barcodes=Tumor,B_cell.
spatialsnake useful_tool --option=splitting results/Colon_Cancer_P2_008um/annotation/Colon_Cancer_P2.zarr --split_by=celltype --barcodes=Tumor,Fibroblast --output_dir=results/useful_results
If --barcodes is not provided, the tool exports one object for every category in the selected field.
Output naming rules:
Without
--barcodes:cluster_<label>.zarrWith
--barcodes:celltype_selected_<label1_label2>.zarrorclusters_selected_<id>.zarr
Scenario 2: split by sample or group
This mode is used to split an integrated object by sample, region, or group.
spatialsnake useful_tool --option=splitting results/merge_data/integrate/concatenated_sdata --split_by=sample --output_dir=results/useful_results
Split by experimental group:
spatialsnake useful_tool --option=splitting results/merge_data/integrate/concatenated_sdata --split_by=group --output_dir=results/useful_results
Output naming rules:
split_by=sampleorregion: export<sample_name>.zarrby coordinate systemsplit_by=group: exportgroup_<group_name>.zarr
Scenario 3: split by ROI CSV from Loupe or Xenium Explorer
How to select ROIs in Loupe
Import the Loupe file generated from Space Ranger into Loupe Browser.
Use the lasso tool to select a region.
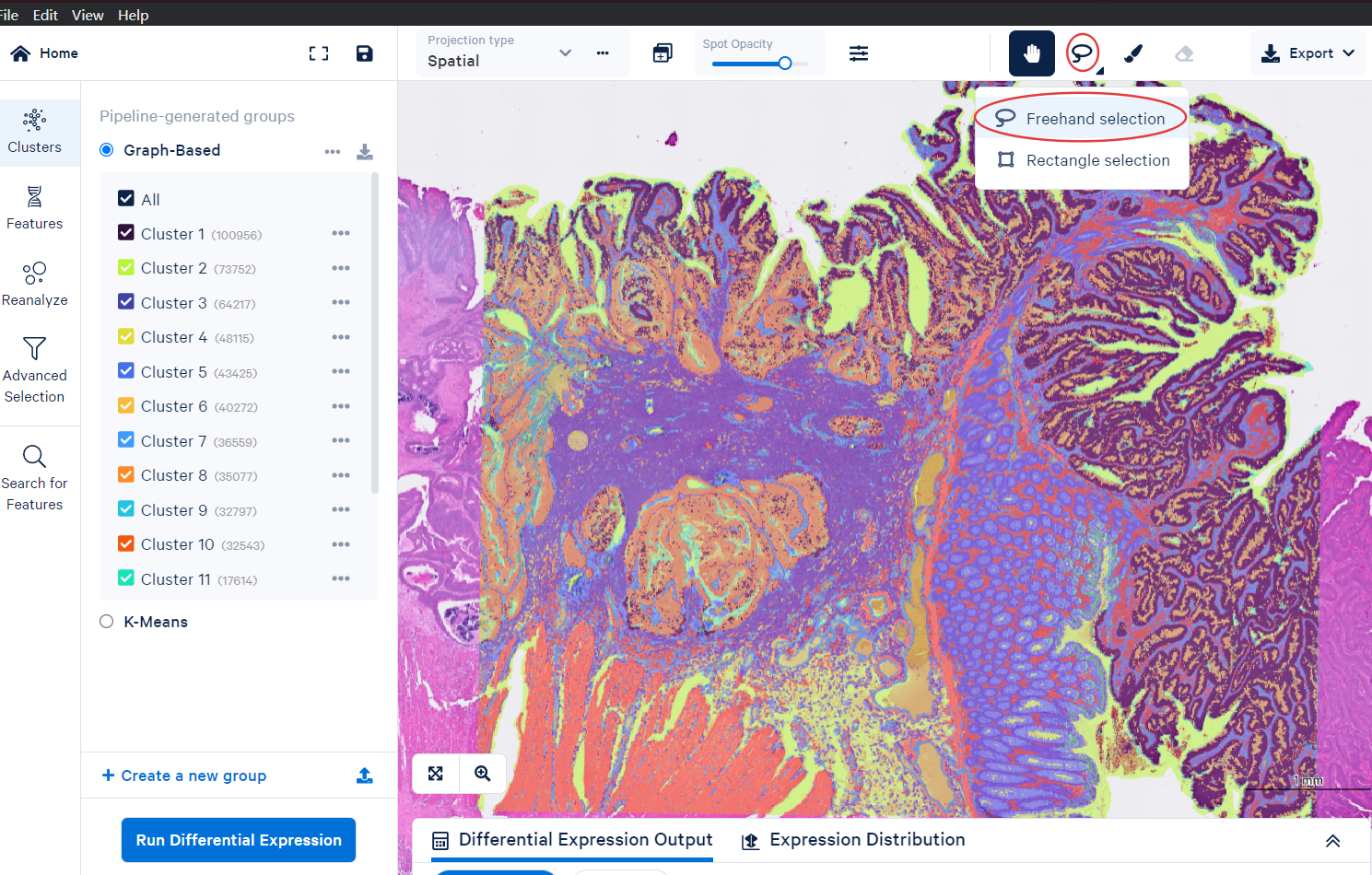
Confirm the region command
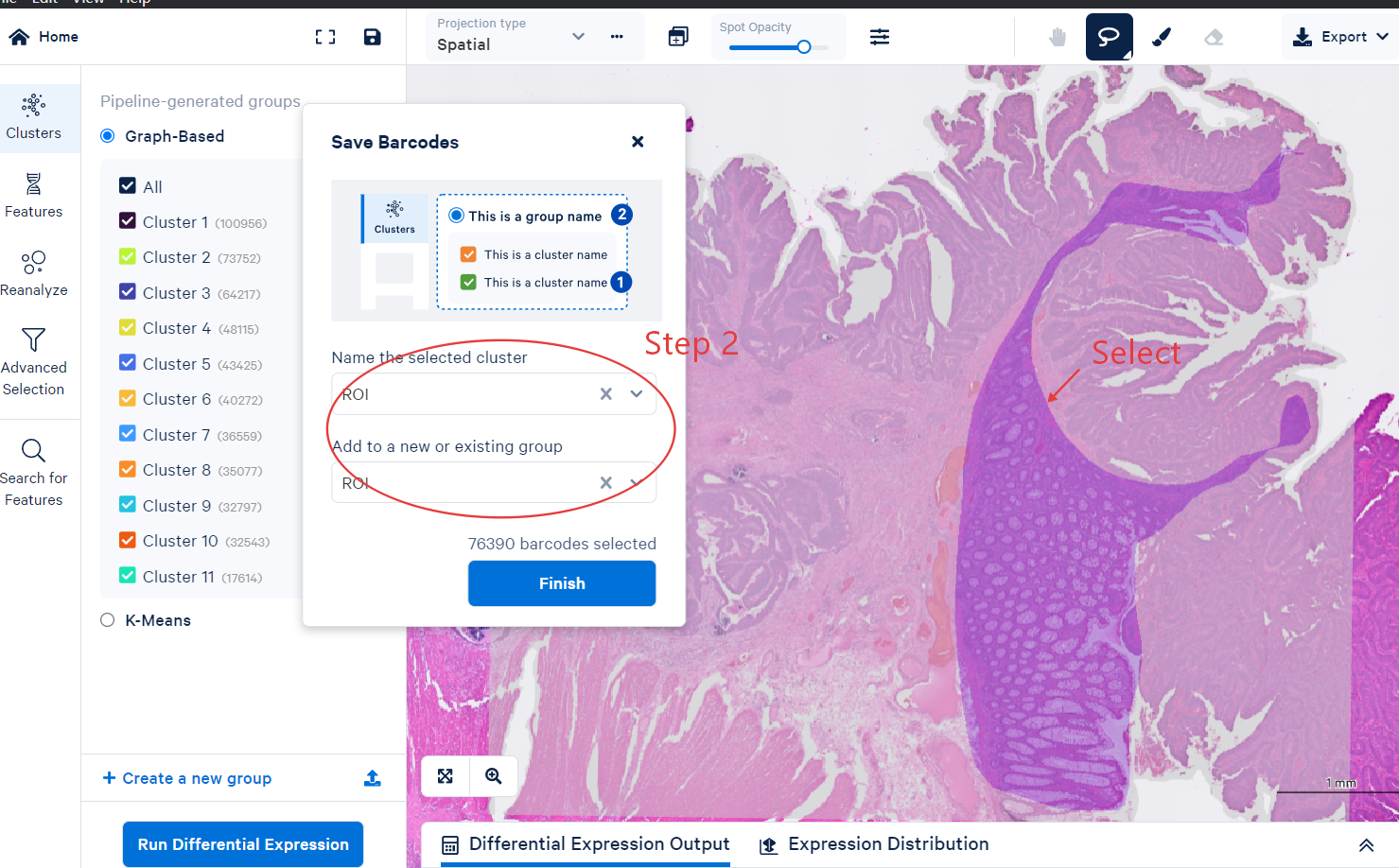
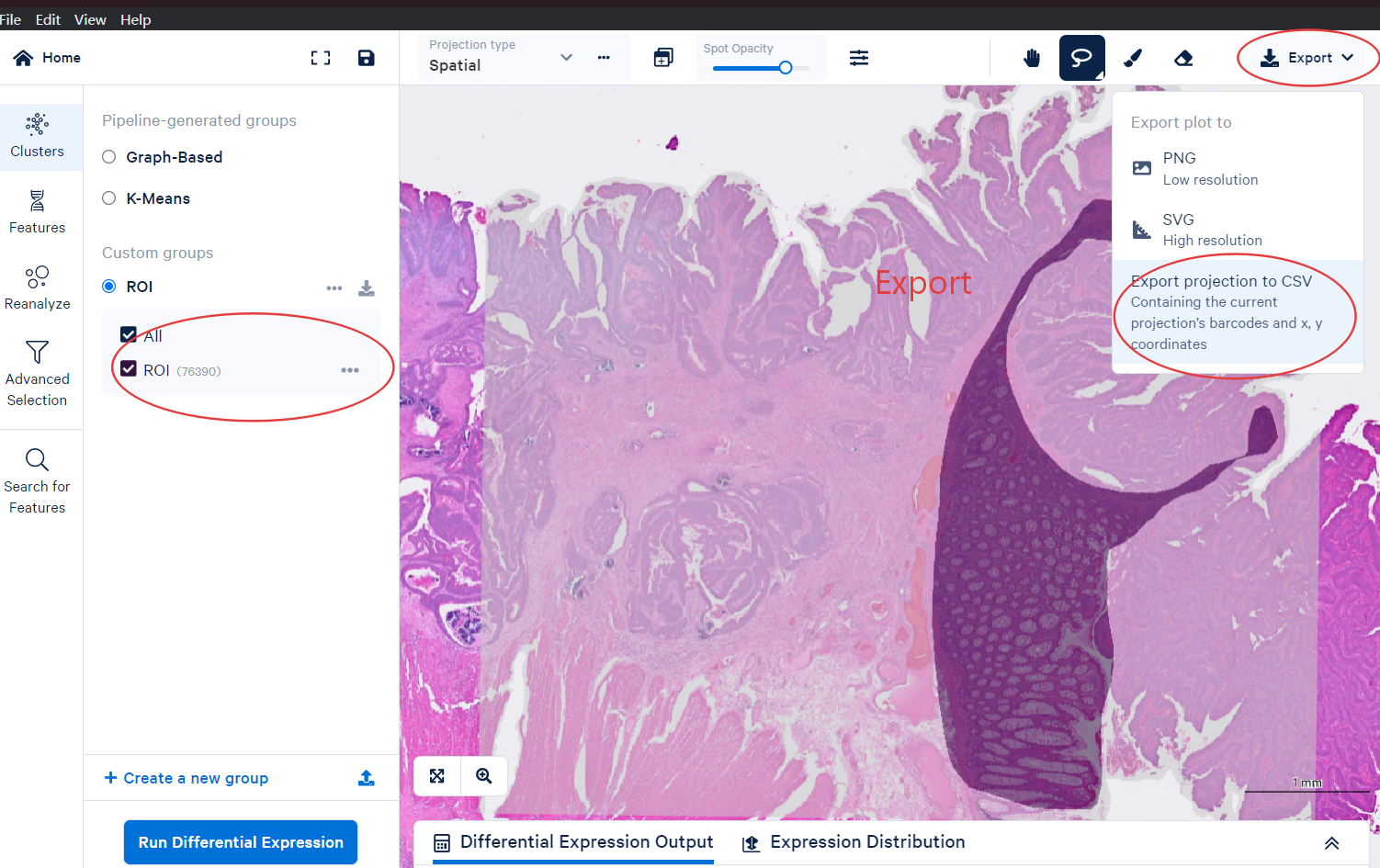
Export the CSV file
How to select ROIs in Xenium Explorer
Import the folder generated by Xenium Ranger into Xenium Explorer.
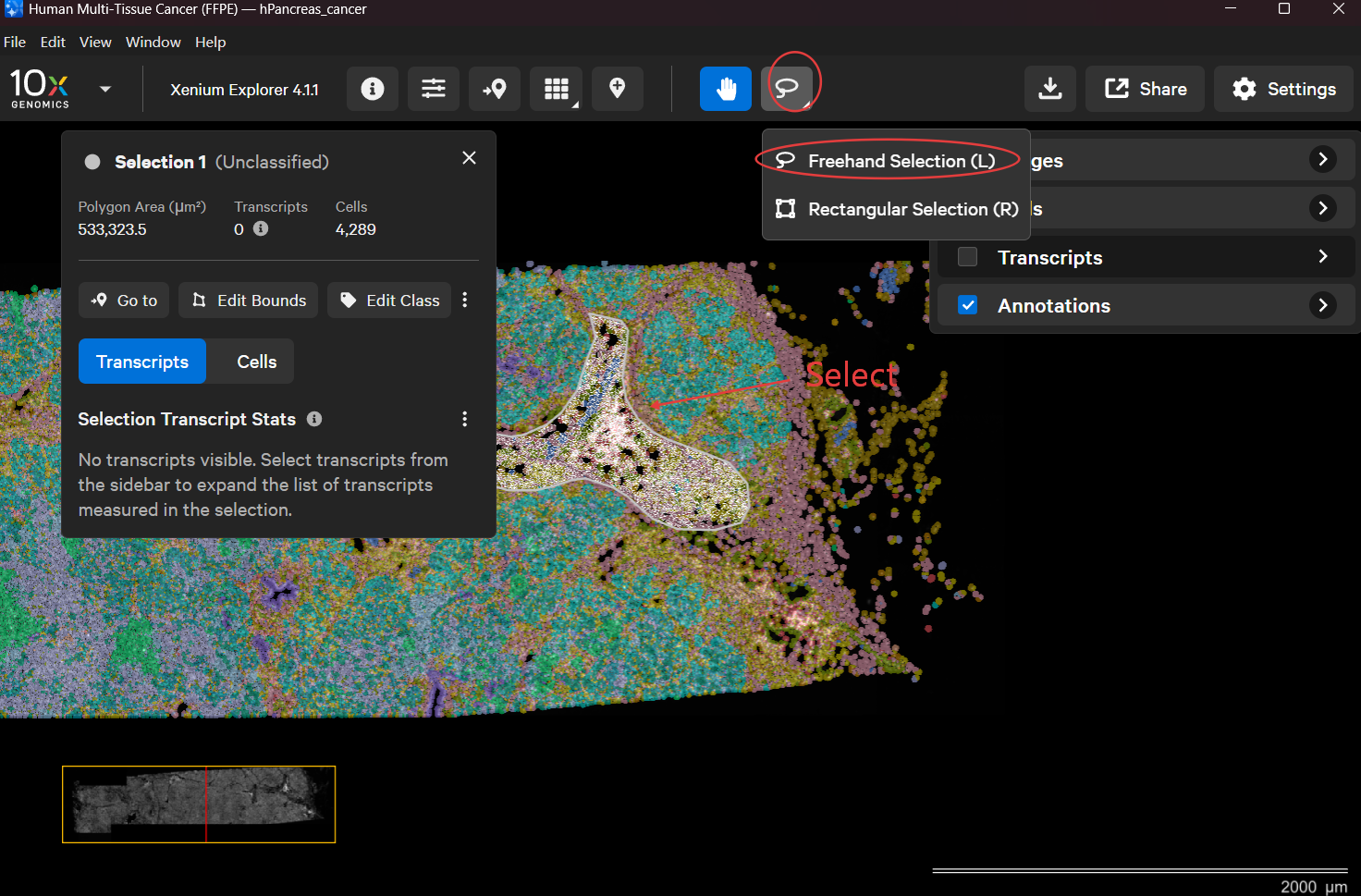
Use the lasso tool to select a region and export it.
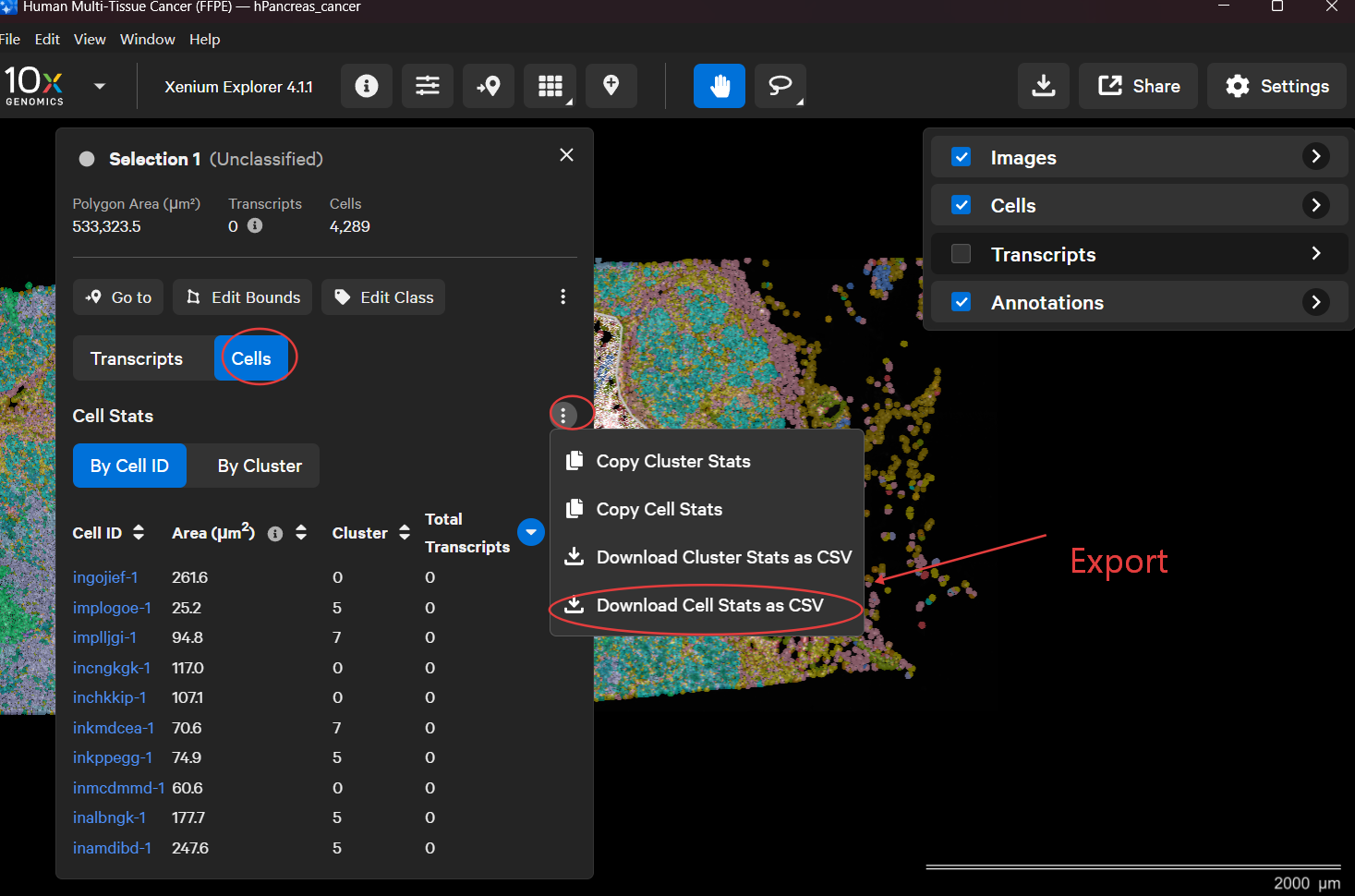
Import csv file for splitting.
spatialsnake useful_tool --option=splitting results/merge_data/integrate/concatenated_sdata --split_by=ROI --roi_csv= [path_to_csv]
roi_csv can be a single CSV file or a directory. If a directory is provided, multiple CSV files inside it are merged automatically.
The CSV file must contain at least a cell_id column. The ROI name column can be roi, region, sample, or group.
If no ROI name column is found, the tool uses the CSV filename as the ROI name.
Output naming rule:
ROI_<ROI_name>.zarr
Scenario 4: crop by image coordinates
This mode crops a tissue region using a coordinate box. The figures generated in annotation_help include coordinate information that can be used as a reference.
This approach is suitable only for rectangular regions. For more irregular or detailed ROI shapes, use Loupe or Xenium Explorer instead.
spatialsnake useful_tool --option=splitting results/Colon_Cancer_P2_008um/annotation/Colon_Cancer_P2.zarr --split_by=image --shape_elements=Colon_Cancer_P2 --min_x=0 --max_x=2000 --min_y=0 --max_y=2000 --output_dir=results/useful_results
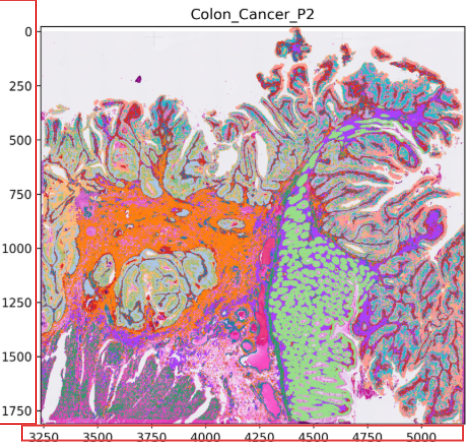
Outputs:
spatial<min_x>_<max_x>_<min_y>_<max_y>.zarr(cropped subobject)<coordinate_id>_shape.png(image plus shape visualization for that region)
Key parameters in practice
Parameter |
Typical values |
Description |
|---|---|---|
|
|
Selects the splitting dimension and is the most important parameter |
|
|
Export only selected categories; if omitted, all categories in the field are exported |
|
|
CSV file or directory used for ROI-based splitting |
|
|
Target layer or sample coordinate system for image-based splitting |
|
|
Coordinate boundaries used for image cropping |
|
|
Output directory for split results |
Common errors and how to fix them
values not found in <field_name>Cause: the values provided in
--barcodesdo not exist in the chosen field.Fix: check spelling and letter case, then confirm whether the correct field is
celltypeorclusters.
group column not found in table obsCause: the object does not contain a
groupcolumn.Fix: use
sampleorregioninstead, or add group information in the upstream workflow.
cell_id column not foundCause: the ROI CSV does not contain a recognizable cell ID column.
Fix: rename the column to
cell_idor another accepted form such asCell IDorbarcode.
Suggested next steps
If you split out a cell population of interest, continue to Secondary Clustering (reclustering) for subcluster refinement.
If you split a multi-sample object, proceed to the downstream modules to compare the biological differences among the resulting subobjects.